

搜索
医疗器械
不良事件监测-MDD警戒系统
撰稿:何丽钦更新日期:2018-01-25
为确保指令的有效实施,欧盟委员会的企业总司发布了《医疗器械警戒系统指南》,为相关方如何切实履行法律义务提供了具体的行动指南。对制造商来说,必须承担两方面的责任:
1. 向主管当局报告不良事件(警戒)
需要报告的不良事件包括:
(1) 死亡或健康状况严重受损:
- 威胁生命的疾病或损害;
- 身体功能的永久性损害;
- 身体结构的永久性损害。
(2) 应会导致死亡或健康严重损害,但侥幸没有发生的事件,或发现器械存在缺陷,这类事件被称为准事件。
制造商提交不良事件报告的时限,即从制造商首次获知事件到成员国主管机构收到通知的时间为:
- 事件:10天;
- 准事件:30天。
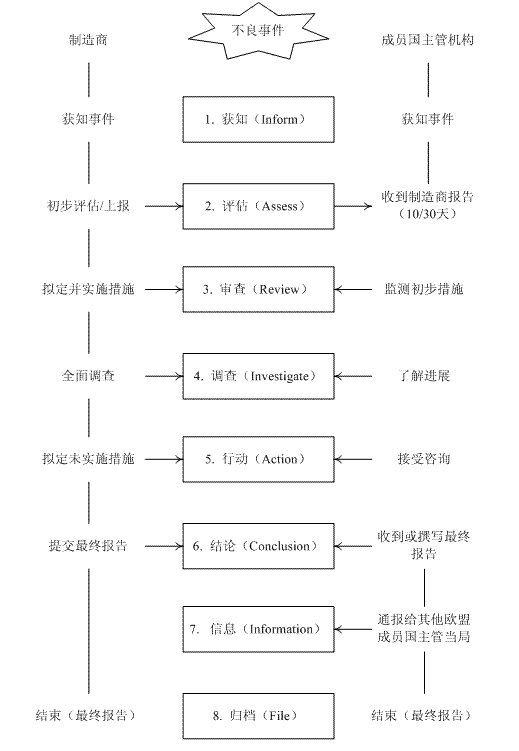
制造商应在最初报告后就不良事件展开调查,并采取必要的措施,包括与主管机构咨询和实施产品召回。同时,制造商还应适时将进展通告。成员国主管机构在限期内,应对调查结果和对相关成员国主管机构处理的反对意见作出书面声明,以最终报告的形式提交。欧盟不良事件调查处理程序如下图所示:
2. 执行一个系统程序,对从市场获得的有关反馈信息建立不良事件档案,并作为质量体系检查的重要部分
内容主要包括:
- 全公司(包括授权代理和经销商)都应了解系统程序的要求;
- 确认向谁报告所有客户投诉和相关不良事件的信息;
- 给予上述确定的人员检验所有投诉和报告,以及做出决定向主管当局报告相应的责任;
- 确认当决定发布劝告通知或产品召回的情况下公司内更高一级人员的责任;
- 指定调查投诉、处理召回产品,以及报告调查结果的程序;
- 保持所有客户投诉、不良事件报告、检验报告和相关信件的程序。
下一篇:确定产品范围



